
来源:X-MOL资讯
多肽分子的体积介于小分子药物与大分子之间,占据着重要的“中分子”(middle molecules)化学生物空间。天然多肽往往需要经过结构修饰才能成为更好的药物分子。在化学修饰技术中,订书肽(peptide stapling)策略在多肽结构和性质的调控以及多肽药物开发上已展示出其独特的功效。然而目前的装订策略往往需要在“处理过”的多肽底物上进行,如引入非天然氨基酸作为特定反应位点或需对侧链高活性基团进行保护。如果能避开底物的预处理,直接对天然多肽进行选择性装订,将会大大拓宽订书肽的适用性,为药物研发提供有力的改造工具。
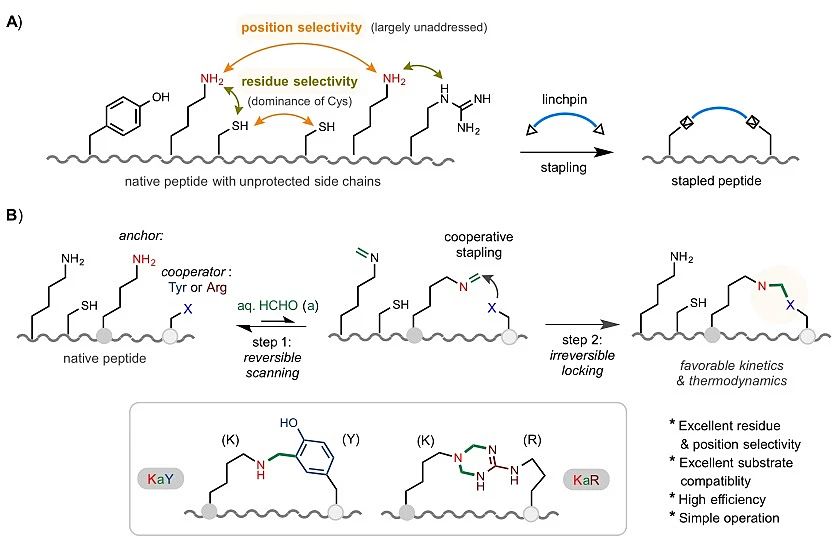
图1. 基于三组分协同反应策略的天然多肽选择性关环
天然多肽上的修饰位点常常被高化学活性的半胱氨酸(Cys)所垄断。此外,现有策略对于多肽上处于不同位置的相同类型残基也基本无法实现精确的位置选择性 (position selectivity) 修饰(图1A)。近日,BETVLCTOR伟德在线登录平台陈弓教授课题组开发了一套非常简单实用的策略,在天然多肽上通过赖氨酸-甲醛-酪氨酸(KaY)或赖氨酸-甲醛-精氨酸(KaR)三组分反应实现了高效高位置选择性成环来构建订书肽。该选择性修饰主要经过两个阶段进行调控:甲醛先通过与底物赖氨酸(Lys, K)侧链上形成可逆亚胺结构来进行扫描;随后周边的酪氨酸(Tyr)或精氨酸(Arg)侧链对亚胺进行分子内亲核进攻“锁定”成最终稳定产物。这个反应过程特有的动力学和热力学特征让该三组分以一种“协同”配合 (cooperative stapling) 的方式实现了基于双组分反应难以实现的位置选择性调控。
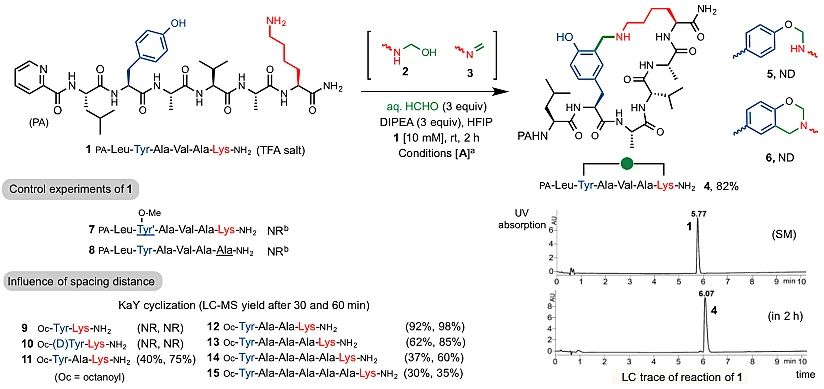
图2. “KaY”多肽关环
考虑到天然多肽具有丰富的亲核性残基,作者设想利用赖氨酸与醛基试剂形成亚胺后再被邻近的亲核基团进攻形成环肽从而实现“订书肽”的构建。作者首先尝试在赖氨酸和酪氨酸之间通过分子内的曼尼希型反应进行成环反应。实验如图2所示,作者发现在室温下以六氟异丙醇(HFIP)为溶剂,模型底物1与极少量的甲醛(3个当量)和碱(DIPEA)在2小时内便可以实现反应的定量转化得到成环产物4,且反应体系非常干净,半缩醛中间体2和3几乎没有产生。HFIP对于该反应的高效转化至关重要,而使用甲醇、乙腈或丙酮之类的溶剂则会明显抑制反应,少量水(<10%)的参与不会对反应影响很大。碱的添加也很重要,通过筛选发现使用有机碱(如DIPEA)收率要高于无机碱(如碳酸钾等)。最终得到的苄胺产物在生理以及酸性和碱性条件下都是可以稳定存在的,作者将这种赖氨酸、酪氨酸以及甲醛的三组分装订反应称为“KaY”反应。随后作者细致考察了赖氨酸和酪氨酸之间的间隔距离对成环反应的影响:通过在两个氨基酸之间增加丙氨酸(Ala)的个数,并在相同时间下来定量分析成环反应的速率。通过1个小时的监测发现,中间没有Ala间隔的底物9未得到任何环化产物,间隔2个或3个Ala的底物显示出较高的反应性(12、13),间隔1个、4个或5 个Ala的底物的反应速度中等。
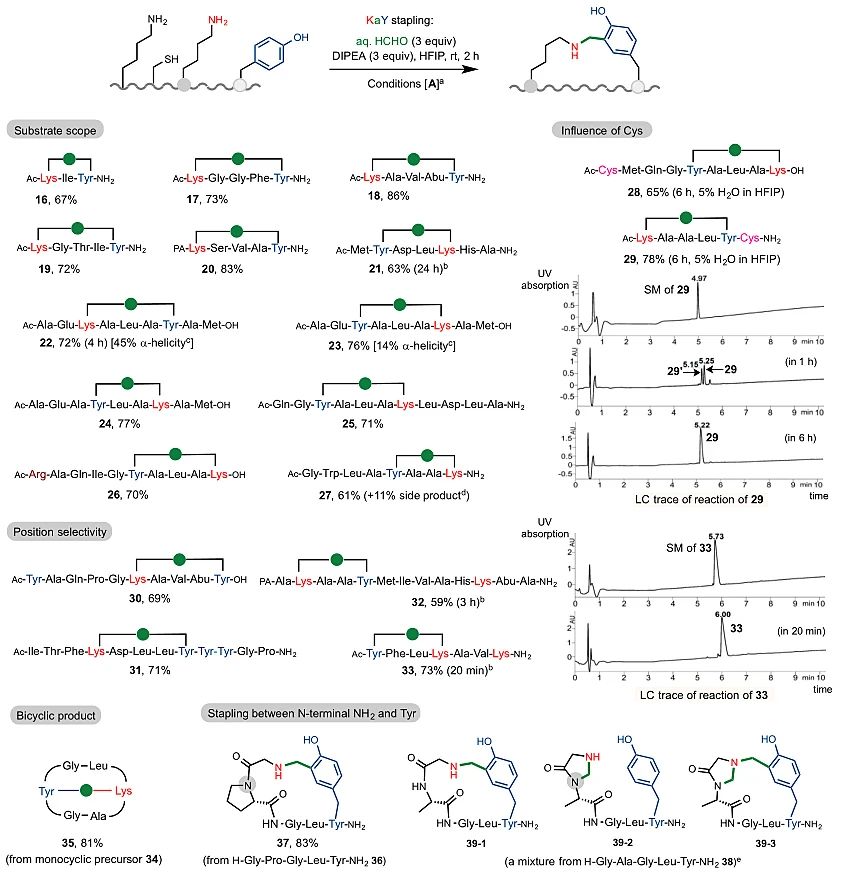
图3. KaY关环的适用范围考察
图3中所示,KaY反应可以兼容多肽上所有的极性基团,如-CO2H(22、25),-CONH2(26),-OH(19、20)等。含有组氨酸的多肽21的成环反应相对较慢,需要延长反应时间并增加甲醛的用量(24小时和5个当量甲醛)。含有色氨酸的多肽会在吲哚环N上产生半缩醛的结构,但并不影响关环反应。带有精氨酸残基的产物26能够以70%的产率得到。含有半胱氨酸的肽链通过对反应位置的调控也能使其不产生干扰(28)。虽然半胱氨酸会很大程度上干扰反应,但是通过实验发现,在含有5%水的HFIP溶剂中,NH-CH2-S键会不稳定,继而释放赖氨酸和半胱氨酸侧链,使这一反应进程被“修正”,朝着生成更为稳定的KaY产物方向进行(29)。随后作者实现了对含有多个相同残基(如酪氨酸或赖氨酸)的肽链进行位点选择性成环修饰。如产物30和31所示,底物中分别含有两个或三个酪氨酸残基时,成环反应将发生在空间上最接近赖氨酸的酪氨酸上;底物中含有两个赖氨酸时,酪氨酸同样将与最近距离的赖氨酸进行成环反应(32和33)。此外,作者通过KaY策略还实现了结构更为复杂的双环肽的构建35。除了侧链-侧链成环,作者还利用N-端游离氨基与侧链进行成环,如37所示,N-端游离的甘氨酸也可以与酪氨酸侧链反应形成环肽产物。但是当N-端第二个氨基酸不是一级氨基酸时,由于末端氨基与相邻氨基酸的酰胺NH之间容易形成稳定的五元缩醛结构从而使得反应变得较为复杂,不易进行分离确认(39-1, 39-2, 39-3)。
在研究不同残基对“KaY”成环带来的干扰时,作者意外发现在相同的反应条件下,精氨酸也能和赖氨酸在甲醛的参与下实现高效成环反应,而目前在温和条件下选择性修饰精氨酸的方法鲜有报道。如图4所示,向HFIP为溶剂的体系中加入6个当量的甲醛,反应2小时后能够以极高产率且选择性地得到以环状四氢-1,3,5-三嗪为特征结构的内外异构体混合物(在LC中的保留时间很近),作者称之为“KaR”产物,相应的反应称之为“KaR”反应。这种三嗪结构产物具有很强的亲水性,这在订书肽策略中也非常少见。
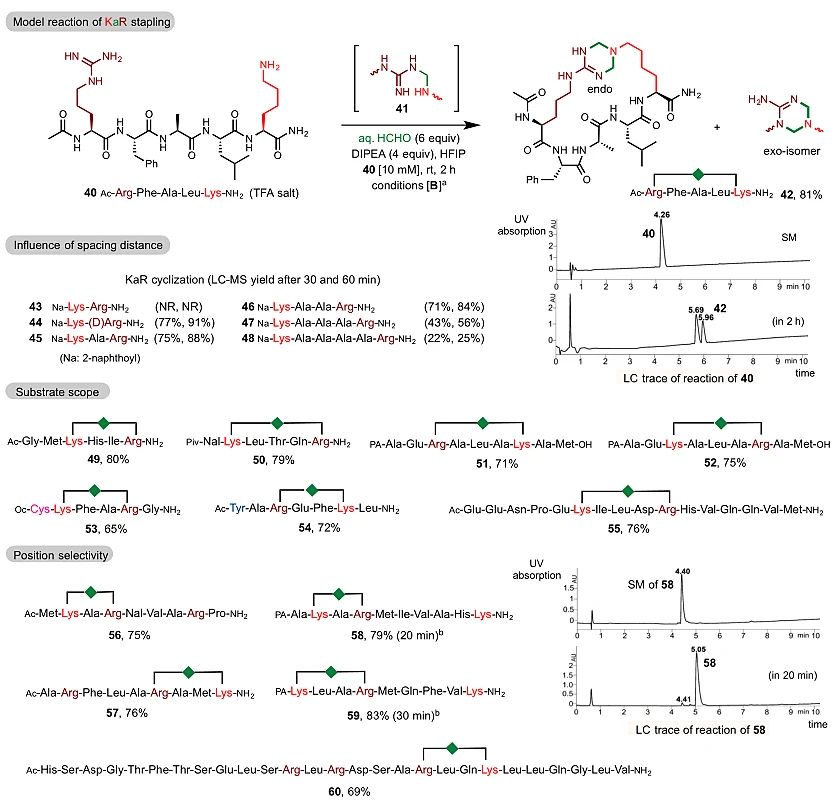
图4.“KaR”多肽关环
与KaY反应相似,彼此相邻的赖氨酸和精氨酸残基之间未发生反应(43),但赖氨酸与相邻D构型精氨酸之间能够以高产率进行成环反应。间隔1或2个丙氨酸的底物在相同条件下显示出最高的反应性(45、46)。在相同条件下,间隔4 个丙氨酸的48成环产率下降至25%。通过作者的观察发现,KaR要比KaY显示出稍高的反应性,并且受其他亲核基团的干扰更小,KaR反应所得到外型和内型异构体的比例很大程度上是受到多肽序列的调控所导致的。在KaR反应中一样,同样可以实现对于包含多个精氨酸或赖氨酸的底物,以高位点选择性进行分子内成环修饰。如产物56和57,底物中有两个精氨酸时赖氨酸将优先与最近的精氨酸成环。带有两个赖氨酸的前体的选择性环化同样会发生在最近的赖氨酸上(58和59)。最后作者还利用KaR策略展示了对二十七肽促胰液素Secretin的高选择性修饰60。
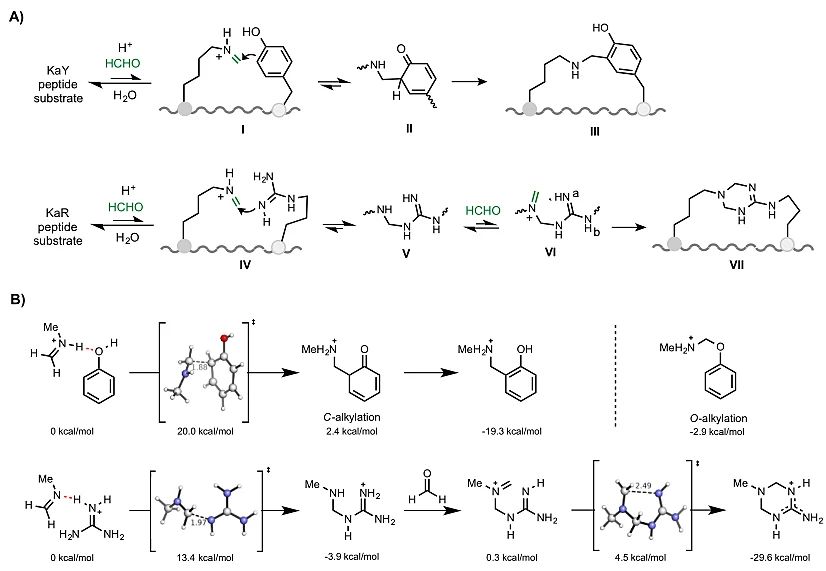
图5. 可能的反应机理及DFT计算研究
随后,通过与UCLA的Ken Houk 教授课题组合作,作者就反应进程通过密度泛函理论(DFT)进行了计算研究(图5B)。由甲醛和甲胺形成的N-甲基亚胺和苯酚来模拟赖氨酸、酪氨酸和甲醛的反应。计算结果表明,苯酚对亚胺离子的亲核进攻是整个过程的限速步骤,能量垒为20.0 kcal / mol。C-烷基化后,接着芳构化释放大量能量,酚类产物形成(∆G = -19.3 kcal / mol)。O-烷基化在热力学上远不令人满意(∆G = -2.9 kcal / mol)。赖氨酸与精氨酸的反应通过胍鎓与N-甲基亚胺的反应进行建模(阳离子亚胺鎓不会直接与胍鎓反应)。该反应过渡态类似于亲核胍进攻N-甲基亚胺基,是模型反应的限速步骤,能垒为13.4 kcal / mol。与甲醛反应并随后环化形成四氢三嗪环非常容易(∆G = 4.5 kcal / mol)。该反应能够释放足够的能量(ΔG= -29.6 kcal / mol),为完成整个反应提供了强大的驱动力。
综上,陈弓教授课题组利用简单的甲醛分子发展了一套新颖和实用的多肽成环策略,以高反应效率和原子经济性实现了在天然多肽上对多个相同残基进行位点选择性的装订修饰,产生了具有独特化学结构的订书肽。这项工作后续将拓展到对更加复杂的多肽甚至蛋白进行选择性修饰,而且还将应用到订书肽分子库的设计中,实现订书肽药物筛选平台的构建。
相关工作最近发表在Angew. Chem. Int. Ed.上,通讯作者为BETVLCTOR伟德在线登录平台的陈弓教授和UCLA大学Ken Houk教授。BETVLCTOR伟德在线登录平台博士研究生李博为该论文的第一作者,硕士研究生唐虹和万照对该工作的顺利进行也做出了重要贡献。
Cooperative Stapling of Native Peptides at Lysine and Tyrosine or Arginine with Formaldehyde
Bo Li, Hong Tang, Aneta Turlik, Zhao Wan, Xiao-Song Xue, Li Li, Xiaoxiao Yang, Jiuyuan Li, Gang He, K.N. Houk, Gong Chen
Angew. Chem. Int. Ed., 2020, DOI: 10.1002/anie.202016267

关注“南开化学”微信公众号