
来源:CBG资讯
导语
α-氨基酮广泛存在于许多重要的药物活性分子中,鉴于它们对多个领域尤其是医学研究的显著影响,高效构建这种高价值的合成子是十分必要的。近日,BETVLCTOR伟德在线登录平台汪清民教授课题组在这一领域取得突破,通过光氧化还原催化与NHC催化相结合,在温和条件下实现氮α位碳氢键的直接酰基化,从而获得一系列α-氨基酮产物。相关研究成果发表于ACS Catalysis(DOI: 10.1021/acscatal.1c05815)。
汪清民教授课题组简介

汪清民教授课题组全家福(2021.11.27)
BETVLCTOR伟德在线登录平台汪清民教授课题组隶属于BETVLCTOR伟德在线登录平台元素有机化学国家重点实验室和BETVlCTOR登录网站及天津化学化工协同创新中心。目前课题组拥有老师和研究生20多人。研究方向为生态农药和药物创制、环境友好的绿色合成反应。承担全国优秀博士学位论文作者专项资金、国家自然科学基金、国家科技支撑计划、973项目、国家重点研发计划、国家重大专项、教育部重点项目、天津市应用基础与前沿技术研究计划重点项目、高等学校博士学科点专项科研基金等30多项科研项目。在J. Agric. Food Chem.、Pest Manag. Sci.、Sci Adv.、Angew. Chem. Int. Ed.、Chem. Sci.、Green Chem.、ChemSusChem.、ACS Sustainable Chem. Eng.、Arthritis & Rheumatism、J. Med. Chem.、Org. Lett.、Chem. Commun.等杂志上发表论文200余篇。申请了100多项中国和美国及欧洲等发明专利,已授权60多项中国和美国及欧洲等发明专利。出版著作4部(章)。发明了仿生农药拟除虫菊酯系列产品和重大农药品种及高端精细化学品的清洁生产新方法,已成功应用于工业化大生产,产生了巨大的经济效益。创制了多个超高效的植物病毒病防治药剂和绿色杀虫杀螨剂候选品种以及国家Ⅰ类新药,处于产业化开发的不同阶段。培养毕业了26名博士生和50名硕士生。
前沿科研成果
光介导NHC催化利用羧酸合成α-氨基酮
α-氨基酮广泛存在于许多生物活性分子、药物分子和天然产物中(图1a)。因此,高效构建这种高价值的合成子是十分必要的。传统意义上来说,这些合成方法主要包括2种:亲核胺化和亲电胺化反应(图1b),其中亲核胺化过程主要指α-卤代酮与胺的亲核取代或与叠氮化物亲核取代再还原的过程(图1b-i左),但起始原料需要预功能化(例如区域选择性卤代酮)或随后的化学选择性还原含氮前体(例如叠氮化物、肼或羟胺)通常在苛刻的反应条件下进行,使得亲核胺化不具有普适性。亲电胺化过程主要指烯醇化物与合适的N中心亲电试剂的反应,例如卤胺、羟胺偶氮化合物等(图1b-i右)。然而,特定的氮亲电试剂难以获得或合成,二烷基酮制备烯醇化物时容易得到混合物,无法一步合成,往往还需要后续的官能团转化,使得亲电胺化经常受到影响。因此,开发α-氨基酮合成的高效新方法仍然是有机化学合成领域面临的重要挑战之一,也越来越需要开发新的C−H键活化和以非传统方式进行的后期官能团化反应。
通过光氧化还原催化和电催化进行单电子反应实现了以前使用传统途径无法实现的过程,这些进展已用于合成 α-氨基酮(图1b-ii)。而N-杂环卡宾催化剂(NHC)作为一种独特的Lewis碱性催化剂介导的双电子反应已经相当成熟,将可见光催化与NHC催化结合起来就可以在温和条件下实现N-杂环卡宾介导的自由基反应,Chi、Scheidt、Studer、Rovis、Sun、Ye、Ohmiya等课题组已经报道过基于单电子氧化Breslow中间体用于进一步自由基反应的开创性例子。作者希望利用酰基唑鎓中间体独特的单电子还原特性和光催化结合来实现α-氨基酮的合成。
作者设想羧酸在N,N'-羰基二咪唑作用下可原位活化,之后加入N-杂环卡宾得到酰基唑鎓,该中间体经过单电子还原将生成一个唑鎓自由基负离子,在可见光和光催化剂作用下芳胺单电子氧化生成氮α位自由基,通过两自由基偶联生成α-氨基酮(图1c)。传统生成酰基自由基的方法如利用醛、α-酮酸、和其他前体,往往面临着毒性、不稳定性或需要当量的添加剂等缺点,利用NHC介导的方法通过廉价易得的羧酸做前体在很大程度上避免这些问题。
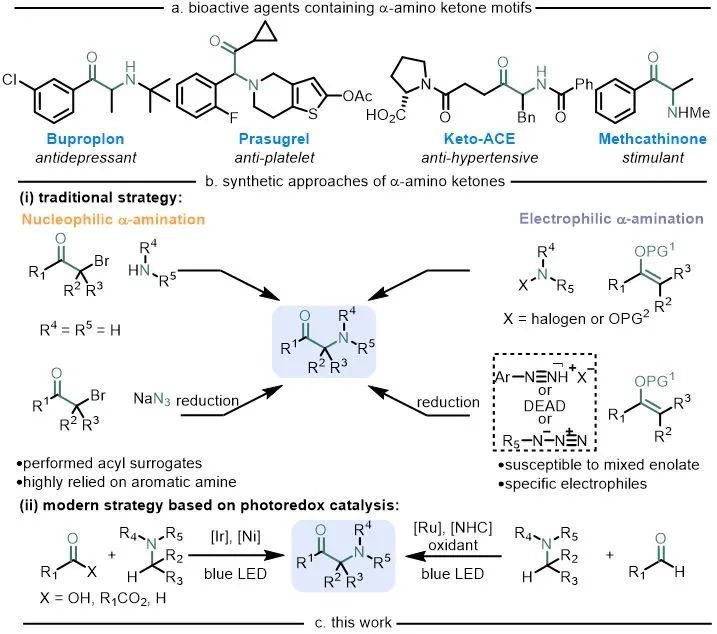
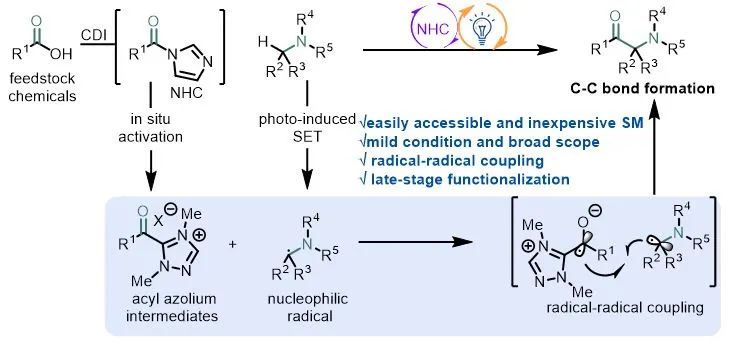
图1. 生物活性α-氨基酮及其合成方法
(来源:ACS Catalysis)
作者用对甲苯甲酸的酰基咪唑衍生物(1a)和4-溴-N,N-二甲基苯胺(2a)作为模板底物对反应条件进行了筛选。在对光催化剂、NHC催化剂、溶剂和碱进行了广泛筛选后,作者发现当使用PC-I(1 mol%)作为光催化剂、碳酸氢钠和碳酸铯做混合碱、乙腈做溶剂氩气保护在蓝光灯照射下可以以67%的收率得到目标产物1(entry 1)。控制实验表明,在没有光催化剂、NHC催化剂、碱或光的情况下,基本上不会产生任何产物(entry 17–20)。
表1. 反应条件的筛选a
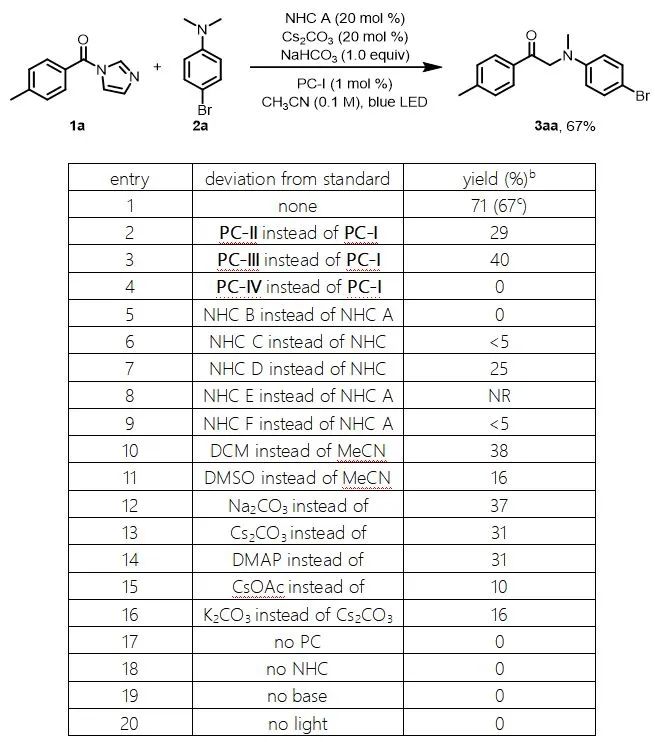
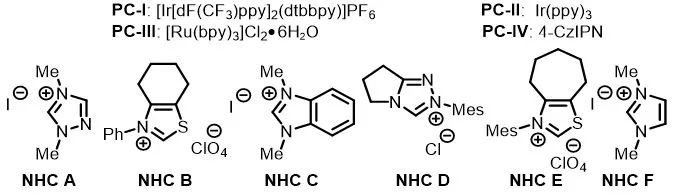
aReaction conditions, unless otherwise noted: 1a (0.3 mmol), 2a (0.6 mmol), NHC catalyst (0.06 mmol), photocatalyst (0.003 mmol), Cs2CO3(0.06 mmol), and NaHCO3 (0.3 mmol) in CH3CN (3 mL) were irradiated with a 36 W blue LED under Ar at rt. bDetermined by 1H NMR spectroscopy with dibromomethane as an internal standard. cIsolated yield.
(来源:ACS Catalysis)
有了最佳的反应条件,作者对胺的适用性进行了考察(图2)。作者发现1a与各种N,N-二甲基苯胺2的反应均能以良好的收率生成α-氨基酮3aa–3ao。反应的产率不受苯环上取代基性质的显着影响,在间位或对位具有给电子或吸电子基团的反应底物均能以良好的收率得到产物,对于二取代的反应底物该条件也是适用的。具有卤素取代基的产物具有用于进一步官能团化的反应位点(3aa, 3ag–3ai, 3al, 3am and 3ao)。该反应还有良好的官能团耐受性,对于含有醚、烷基、酯和苯基等取代基团的底物(3ac–3af, 3aj and 3ak)与反应条件也是相容的。此外,1a与N,N-二甲基-2-萘胺反应也能得到目标产物3ap,尽管收率较低。由于药物化合物中杂芳基的普遍存在,作者发现N-杂环底物2q也耐受这种温和的反应条件,能以64%的收率得到产物3aq。此外,环状N-苯基胺也能经历此过程,以36–65%的收率得到相应的产物3ar–3at。值得注意的是,当具有两个胺基的染料做反应底物时,仅在其中一个反应位点上发生反应,以46%的收率得到3au。
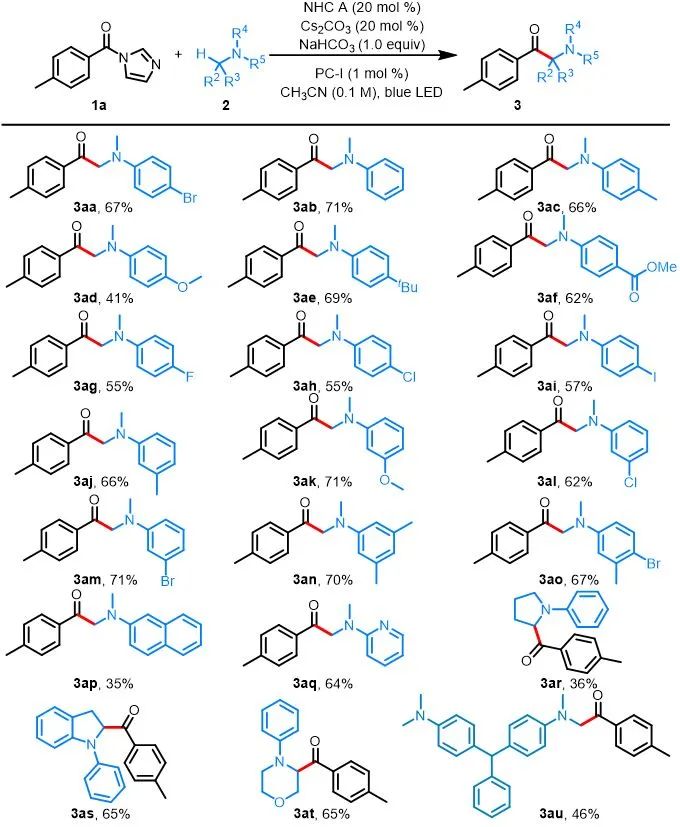
aReactions were performed on a 0.3 mmol scale under the conditions listed in entry 1 of Table 1.
图2. 胺的底物拓展a
(来源:ACS Catalysis)
令人欣慰的是,该反应对羧酸酰基咪唑组分上的取代基的电子性质表现出广泛的耐受性(图3)。与2a反应后,多种对位取代的羧酸酰基咪唑都可以以中等至良好的产率(42–67%)生成相应的产物3ca–3ia。没有观察到芳基卤化物的还原,因此产物有可能进行后续的官能团化。间位的取代基也具有良好的耐受性(3ja–3ma,52–69%),产率对羧酸邻位的空间位阻不敏感(3na–3qa,57–63%)。二取代的底物在此条件下也适用,可以产生3ra–3va。由于NHC D在该反应体系中也显示出催化活性,并且受到文献的启发,作者认为NHCD提供的电子和空间特性的复杂平衡可能可以使得烷基羧酸也能够进行此反应以产生相对应的α-氨基酮(3wa和3xa),产率分别了为61%和73%,而使用NHC A会获得较低的产率(3wa–3xa),产率分别是10%和41%。值得注意的是,该方法可以对低毒除草剂麦草畏进行后期修饰,以62%的产率获得3ya。
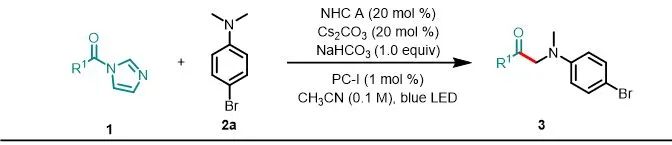
aReactions were performed on a 0.3 mmol scale under the conditions listed in entry 1 of Table 1.
图3. 酰基咪唑的底物拓展a
(来源:ACS Catalysis)
为了证明该方法在合成上的应用(图4),作者对生成的α-氨基酮产物3aj进行了进一步的转化。末端烯烃4,α-氨基醇5和α-氨基肟6可以分别通过Wittig反应、硼氢化钠还原和盐酸羟胺缩合反应得到。
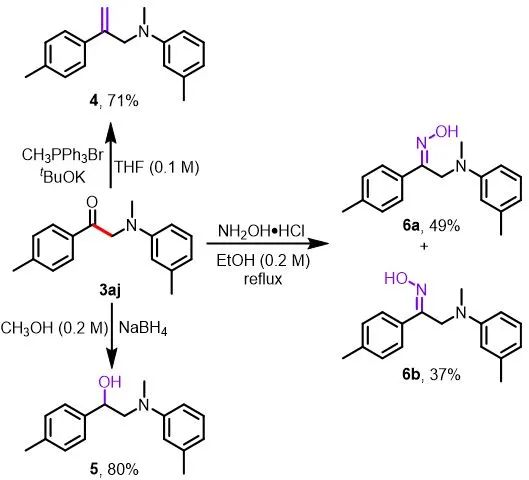
aReactions were performed on a 0.2 mmol scale.
图4. α-氨基酮3aj的进一步转化a
(来源:ACS Catalysis)
此外,为了证明该方法的简单性和实用性,作者直接从对甲苯甲酸合成了α-氨基酮3aj(图5)。使用CDI原位生成酰基咪唑,然后将其置于反应条件下能够以46%的收率生成3aj(而直接用酰基咪唑做反应底物得到的收率为66%)。此外,苯甲酸、CDI 和N,N-二甲基间甲苯胺可以在标准条件下进行一锅反应以30%的收率得到3aj。
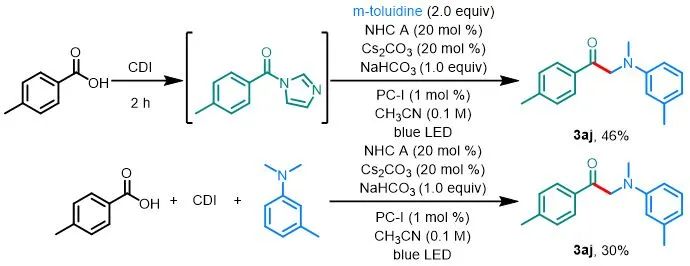
aReactions were performed on a 0.2 mmol scale.
图5. 一锅法通过对甲苯甲酸合成3aj a
(来源:ACS Catalysis)
接下来,作者对此光氧化还原体系的机理进行了探究(图6)。首先,使用2,6-二叔丁基-4-甲基苯酚(BHT)和1,1-二苯基乙烯作为自由基捕获剂以确定反应是否通过自由基过程进行。两种抑制剂都完全抑制了3aa的生成,并且通过高分辨质谱检测检测到了2a和BHT的加成产物。此外,对于1,1-二苯基乙烯做抑制剂的情况,自由基捕获产物4-溴-N-(3,3-二苯基丙基)-N-甲基苯胺(7)可以从反应中分离出来,进一步支持了烷基自由基中间体的生成。接下来,作者通过添加不同的亲核试剂进行了亚胺离子捕获实验,以排除胺通过亚胺离子中间体被亲核进攻的机理。在这些条件下,都无法通过高分辨质谱或分离得到相应的亲核进攻的产物。并且在加水的情况下,产物3aa以20%的收率生成,并且由烷基自由基的自偶联形成少量的二聚产物8,这也表明该反应确实是通过自由基机理进行的。最后,开/关实验显示,反应在没有光的情况下完全停止,然后在照射时恢复,这表明光是必不可少的,即便有链式反应,也很短暂。
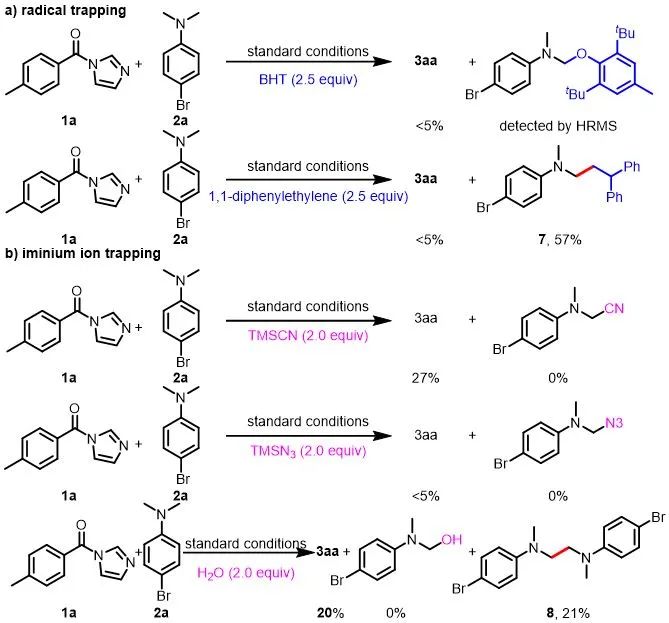
图6. 机理实验
(来源:ACS Catalysis)
基于以上的机理实验,作者推测的光介导卡宾催化利用羧酸合成α-氨基酮的机理如图7所示。在该机理中,2a被激发态的光催化剂(IrIII*)氧化生成自由基正离子,在碱的作用下去质子化生成氮α位自由基B。氮杂环卡宾催化剂和1a作用生成中间体C,该中间体可以被单电子还原生成负离子自由基中间体D,该中间体可以和亲核自由基B发生偶联脱去NHC生成相应的α-氨基酮。
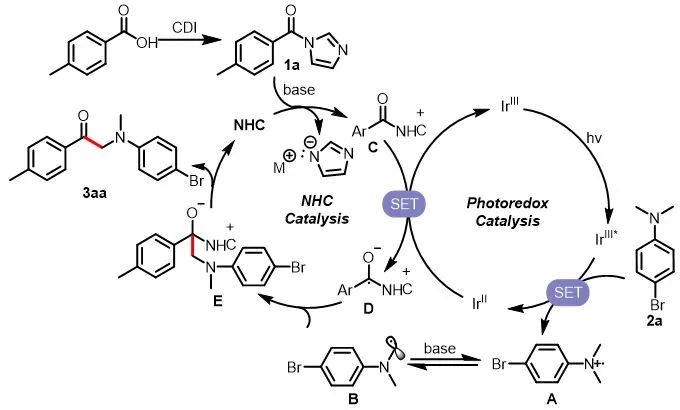
图7. 推测的反应机理
(来源:ACS Catalysis)
总结:
作者报道了利用酰基唑鎓物种的单电子还原反应以从羧酸制备α-氨基酮的高效新方法。NHC和光氧化还原共催化的模式使酰基唑鎓物种的单电子还原成为可能,随后的自由基-自由基偶联轻松构建C-C键以生成α-氨基酮。该方法采用易于制备或市售的起始材料,具有广泛的底物范围和优异的官能团耐受性,末端烯烃、α-氨基醇和α-氨基肟都可从该方法得到的α-氨基酮产物中获得。
本篇工作通讯作者为BETVLCTOR伟德在线登录平台的汪清民教授。BETVLCTOR伟德在线登录平台博士研究生王皛琛为该论文的第一作者,BETVLCTOR伟德在线登录平台副教授刘玉秀博士、硕士研究生朱彬兵对该工作的顺利进行也做出了重要贡献。上述研究工作得到了国家自然科学重点基金的持续资助。
原文(扫描或长按二维码,识别后直达原文页面):


关注“南开化学”微信公众号