
来源:DFT代算
电催化将CO2转化为甲酸盐是公认的一种经济可行的CO2升级途径,但由于驱动相关质子耦合电子转移(PCET)过程的能垒较高,以及对第二个PCET的严重忽视,因此需要较高过电位才能实现高选择性。
2024年6月13日,BETVLCTOR伟德在线登录平台王欢研究员/程方益研究员、吴金雄研究员团队合作在Angew. Chem. Int. Ed.期刊发表题为“Sequentially Regulating Potential‐Determining Step for Lowering CO2 Electroreduction Overpotential over Te‐Doped Bi Nanotips”的研究论文,BETVLCTOR伟德在线登录平台李有增、李金翰、艾威为论文共同第一作者,吴金雄研究员、程方益研究员、王欢研究员为论文共同通讯作者。
该研究通过对Te掺杂Bi(TeBi)纳米尖端的电位决定步骤(PDS)进行顺序调节来克服这一挑战。计算研究揭示了Te杂原子的加入通过大幅降低*OCHO中间体的形成势垒,改变了从第一个PCET到第二个PCET的PDS,而高曲率纳米尖端可诱导增强的电场,从而引导不对称*HCOOH的形成。在这种情况下,*OCHO和*HCOOH的热力学势垒可以依次降低,从而在低过电位下实现高甲酸选择性。在实验中,通过控制前驱体中Te的含量可以获得不同的TeBi纳米结构,在相对正的电位窗口(-0.57V至-1.08V)内,TeBi纳米尖端生产甲酸盐的法拉第效率>90%。强Bi-Te共价键还具有很强稳定性。在优化的膜电极组装装置中,3.2V时的甲酸盐产率达到10.1mmol·h-1·cm-2,显示出巨大的实际应用潜力。

DOI:10.1002/anie.202407772
基于铋(Bi)催化剂的独特优点,如低毒性、地球丰产性、对析氢反应(HER)的惰性等,研究人员提出了一种巧妙的策略,即在Te掺杂Bi(TeBi)纳米尖端顺序调节CO2转化为甲酸盐的PDS,从而在正电位窗口内获得高甲酸盐选择性。密度泛函理论DFT计算显示,Te杂原子的引入不仅极大稳定了*OCHO 中间体,改变了第二个PCET的PDS,而且还调节了不对称*HCOOH中间体的取向,增加了偶极矩 (µ)。有限元FEM仿真模拟结果表明,高曲率纳米尖端的构建会诱发局部增强的电场(E),从而产生更强的µ-E相互作用,并稳定*HCOOH中间体的形成。因此,在Te掺杂Bi的纳米尖端上,CO2转化为甲酸盐的热力学势垒大大降低,从而有利于甲酸盐在更低的过电位下形成。实验中,通过控制前驱体中Te的含量,制备了不同曲率半径的Te掺杂Bi纳米结构,高曲率Te掺杂Bi纳米尖端在-0.57V至-1.08V的电压范围内表现出卓越的CO2到甲酸的选择性,法拉第效率高于90%。此外,Bi和Te具有相似的电负性,从而产生了较强的Bi-Te共价关系,实现了CO2到甲酸盐的稳定转化。在膜电极组件(MEA)装置中,Te掺杂Bi纳米尖端通过耦合5-羟甲基糠醛的氧化,在3.2 V的电池电压下提供了542.0mA·cm-2的偏电流密度。该研究强调了调控第二个PCET的重要性,并为设计高效甲酸选择性电催化剂提供了一条新途径。
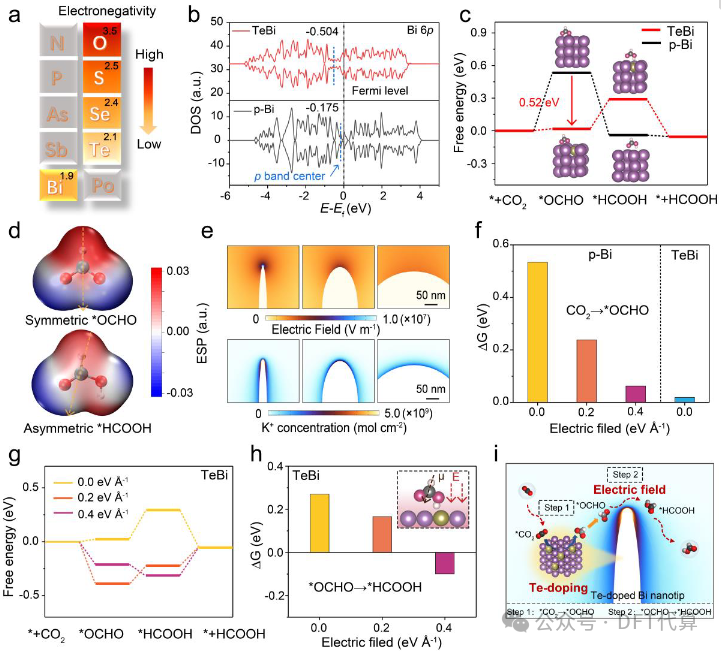
图1. DFT计算和有限元模拟。(a) O族元素与金属Bi的电负性差异。(b) p-Bi和TeBi催化剂中Bi元素p轨道的态密度。(c) p-Bi和TeBi表面甲酸生成的自由能分布图。插图:*OCHO和*HCOOH在催化剂表面的吸附结构。(d) *OCHO和*HCOOH中间体的静电势(ESP)分布。(e) Bi NTs上的电场和K+浓度分布。(f)不同电场强度下TeBi和p-Bi表面的ΔG*OCHO。(g) 不同电场下TeBi表面产生甲酸盐的自由能图。(h) 不同电场下TeBi表面上的ΔG*HCOOH。插图:*HCOOH 中间体的相应吸附构型。(i) 提议的TeBi NT上CO2RR的反应机制。紫色和棕色球分别代表Bi和Te。
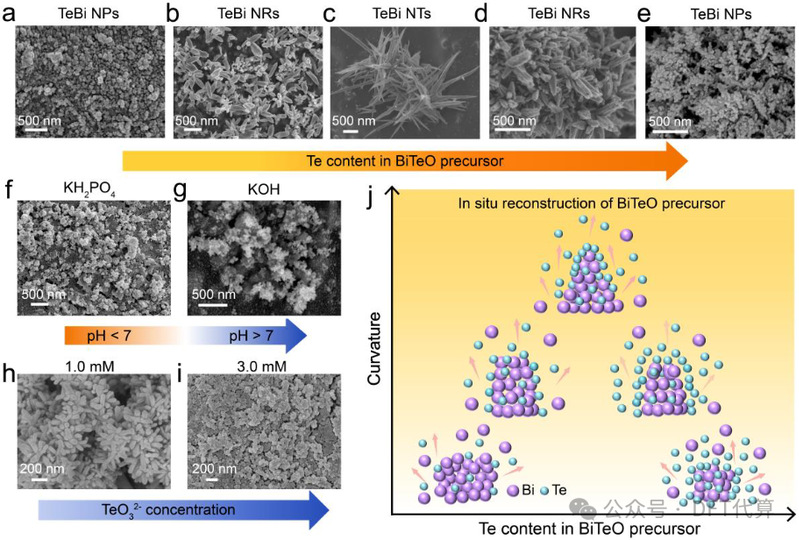
图2. 不同Te含量的BiTeO衍生出的Bi纳米结构的合成与表征。(a-e) 通过电还原不同Te含量的BiTeO化合物得到的TeBi NRs、TeBi NTs和TeBiNPs的SEM图像。BiTeO-Te7.0在 (f) KH2PO4电解质(pH<7)和 (g) KOH电解质(pH>7)中电还原后的SEM图像。在加入 (h) 1.0mM TeO32- 和 (i) 3.0mM TeO32- 电解液的KHCO3电解液中进行电还原后BiTeO-Te7.0的SEM图像。(j) 不同Te含量的BiTeO形成TeBi NRs、TeBi NTs 和 TeBi NPs的重构过程示意图。
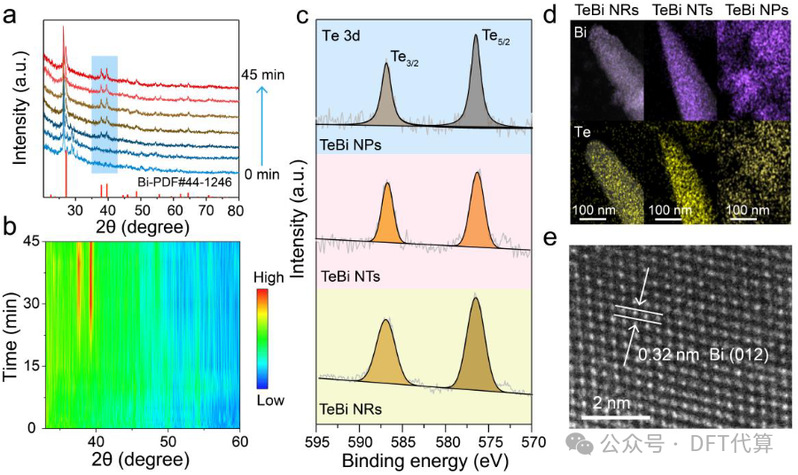
图3. 催化剂的表征。(a)用于监测BiTeO转化为TeBi NTs的原位XRD图。(b)相应的等值线图。TeBi NRs, TeBi NTs和TeBi NPs的 (c) 3d XPS光谱和 (d) EDS映射。(e) TeBi NTs的HRTEM图像。
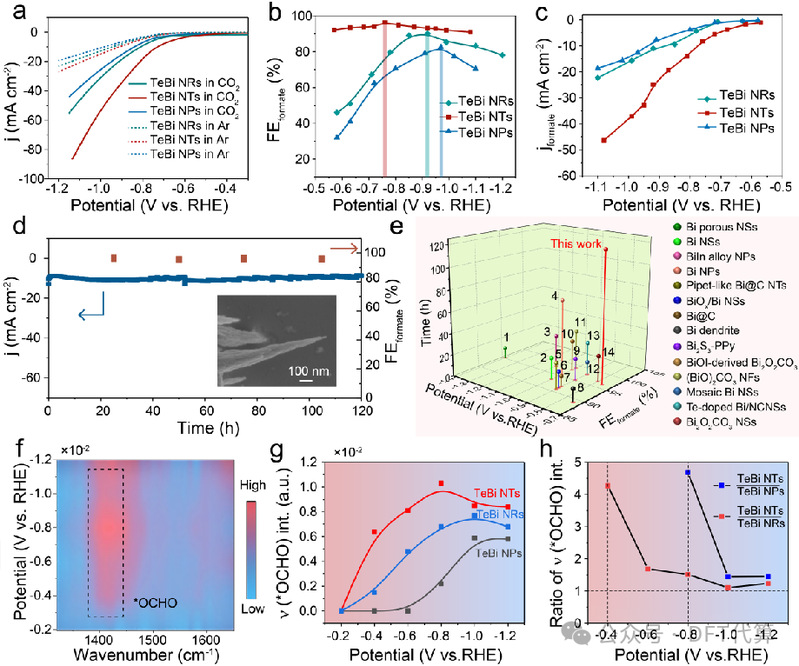
图4. CO2电还原性能研究。(a) 不同电位下TeBi NRs、TeBi NTs和TeBi NPs的LSV曲线、(b) FEformate 和 (c) jformate。(插图:长期CO2RR后TeBi NTs的SEM图像。(e) TeBi NTs在H型电池中产生最大FEformate电位下的耐久性与最近报道的大多数Bi基电催化剂的耐久性的比较。(f) TeBi NTs上原位ATR-SEIRA的等值线图。(g) 从TeBi NRs、TeBi NTs和TeBi NPs原位ATR-SEIRA光谱中获得的 ν (*OCHO) 峰强度 (int.) 和 (h) 峰比。
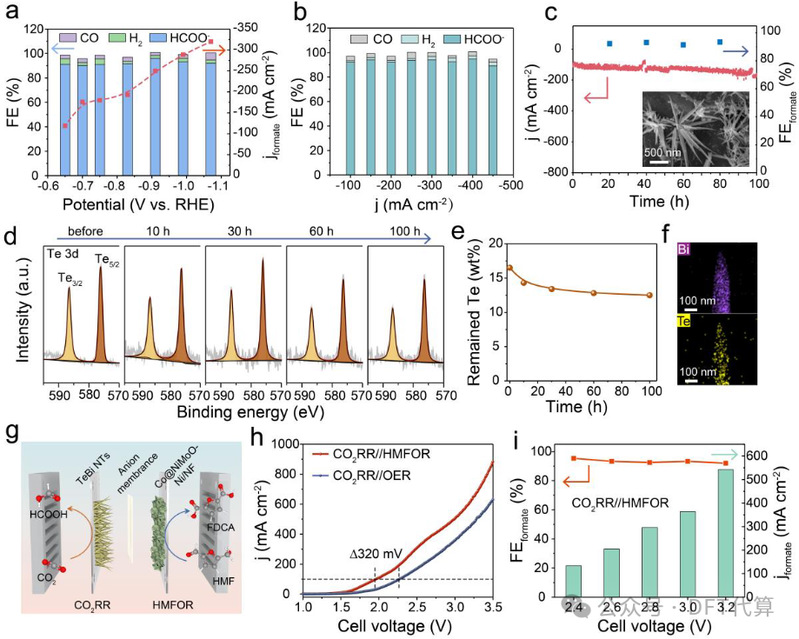
图5. 液流电池和MEA电池测试。(a)流动池中不同电位下的FEformate和jformate。(b)流动池中不同电流密度下的FEformate。(c) TeBi NTs在CO2RR液流电池中-0.65 V下的长期时变稳定性。插图:TeBi NTs在流动池中CO2RR 100小时后的SEM图像。(d)不同CO2电还原时期TeBi NTs的Te 3d XPS光谱。(e)相应的Te含量。(f) CO2RR后TeBi NTs中Te和Bi的EDS映射。(g) MEA电池配置中的CO2RR//HMFOR耦合系统示意图。(h) MEA电池中CO2RR//HMFOR和CO2RR//OER系统的LSV曲线。(i) CO2RR//HMFOR系统在不同电池电压下的FEformate和jformate。
总之。在密度泛函理论DFT计算和有限元FEM模拟的指导下,通过优化前驱体中的Te含量,制备了Te掺杂的Bi纳米尖端,从而实现了PDS的顺序调节,以较低的过电位实现了高选择性的甲酸生产。计算研究表明,Te杂原子的引入显著降低了*OCHO的形成能垒,使得形成*HCOOH的第二个PCET成为CO2转化为甲酸盐的PDS。有限元FEM模拟结果表明,在纳米尖端周围产生了局部增强的电场,使得不对称的*HCOOH中间体得到极大的增强。因此,CO2转化为甲酸盐的热力学势垒大大降低。实验结果表明,在一个相对正的电位窗口(-0.57V至-1.08V)内Te掺杂的Bi纳米尖端生产甲酸盐的法拉第效率>90%,优于TeBi NRs(-0.92V时为90.2%)和TeBi NPs(0.97V时为82.6%)。此外,Bi和Te的电负性相近,形成了牢固的Bi-Te共价键,从而在CO2RR过程中将Te锁定在Bi晶格中,从而实现了超过120小时连续运行的卓越稳定性。在与优化氧化反应耦合的MEA电解槽中,甲酸盐产率达到10.1mmol·h-1·cm-2,电池电压仅为3.2V。该研究工作为促进CO2电还原生成甲酸盐提供了一种新的催化机制。

关注“南开化学”微信公众号